
安渡视点 | 一文了解中美欧药品注册沟通交流会议
本期为大家介绍中美欧药品注册沟通交流会议。
 \
\
▎本文作者为安渡生物药政事务团队
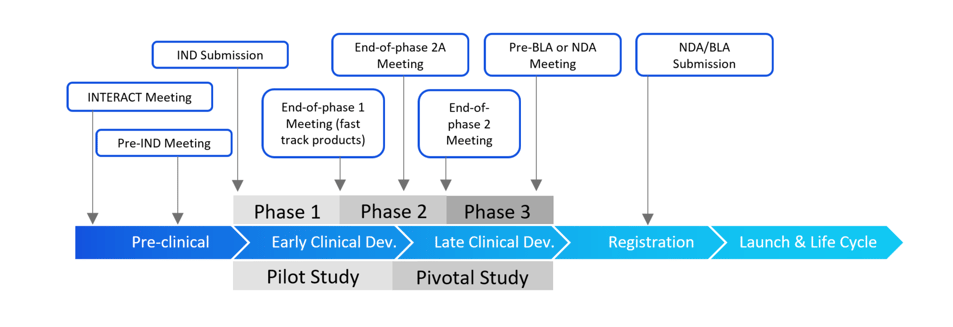
中美欧药品注册沟通交流会议介绍如下:
![]() 中国
中国
-
召开时限:Ⅰ类会议一般安排在申请后30个工作日内。
-
包括的会议类型:新药临床试验申请前会议(Pre-IND);药物Ⅰ期临床试验结束/药物Ⅱ期临床试验结束(或启动前)/Ⅲ期临床试验启动前会议(EOP1/EOP2);新药上市许可申请前会议(Pre-IND/BLA);风险评估和控制会议;申请附条件批准和/或适用优先审评审批程序的沟通会议 -
召开时限:Ⅱ类会议一般安排在申请后60个工作日内。
-
包括的会议类型:新增适应症及新增药物联用的临床申请;讨论临床急需或罕见病药物药法过程中的重要问题;讨论复杂仿制药、一致性评价或再评价品种的重大研发问题;讨论复杂的重要非临床研究的设计方案;讨论与药审中心的技术分岐;前沿技术领域药物的讨论;上市后的变更相关问题的讨论;临床试验期间,对于安全性评估及风险管理存在问题的讨论;讨论上市后临床试验设计。 -
召开时限:Ⅲ类会议一般安排在申请后75个工作日内。
![]() 美国
美国
-
包括的会议类型:21CFR10.75、312.48和314.103中提及的争端解决会议;讨论药物开发中的临床暂停情况,并着眼于讨论新的开发方向的会议;在收到 FDA 根据特殊临床方案评估程序的评估后,申办方要求召开的特殊临床方案评估会议;在FDA做出监管行动(批准除外)的3个月内要求召开的Post-action会议;在FDA发出拒绝申请信函(refuse-to-file letter)后30天内要求召开的会议。 -
会议时限:FDA会议安排时间为收到申请后30个日历日内。
-
包括的会议类型:Pre-IND会议;Pre-NDA会议,Pre-EUA会议;审查综合疗效总结 (ISE) 和综合安全性总结 (ISS)的会议;在 FDA 做出监管行动(批准除外)3 个月以后要求召开的Post-action会议;在上市申请审查范围之外的,针对产品风险评估和应对策略或上市后要求的会议;讨论获得突破性疗法认证的产品的整体开发计划会议。 -
会议时限:Pre-IND FDA响应时间21个日历日,FDA会议安排时间为收到申请后60个日历日内;Pre-NDA/BLA响应时间14个日历日,FDA会议安排时间为收到申请后70个日历日内。
-
包括的会议类型:EOP1针对治疗危及生命和严重衰竭性疾病的生物制品和获得加速批准并用于严重/危及生命的的药物;EOP2/3。 -
会议时限:响应时间14个日历日,FDA会议安排时间为收到申请后70个日历日内。
-
包括的会议类型:EOP2A会议;OTC 各论反馈会议;Phase 4会议。 -
会议时限:FDA响应时间21个日历日,FDA会议安排时间为收到申请后75个日历日内。
-
包括的会议类型:在正式会议中提出的新议题的相关后续问题;针对一个仅包含数个问题的小规模议题寻求监管机构建议;关于尖端创新研发方向的问题。 -
会议时限:响应时间14个日历日,FDA会议安排时间为收到申请后50个日历日内。
-
会议时限:FDA响应时间21个日历日,FDA会议安排时间为收到申请后90个日历日内。
-
针对仿制药开发过程的GDUFA Scientific Meetings,包括ANDA申请递交前、开发中和上市审批过程中的不同会议类型; -
针对医疗器械开发过程的MDUFA Meetings,包括Q-Sub会议; -
针对新靶点、全新化合物以及临床数据不完整而设计的Advisory Committee Meetings,比如ODAC。
-
SA会议的目的:为申办方药品开发的具体问题提供科学建议。 -
SA会议涉及的问题:CMC方面、药理毒理方面、临床方面、统计学方面、整体开发策略、孤儿药认定以及儿科开发计划等。 -
SA会议时限:SAWP(科学建议工作组)每月定期召开会议讨论申请人的问题,EMA每年会公布年科学建议小组会议时间,申请人要按照所公布时间及时提供会议申请。 -
SA会议费用:EMA对科学咨询收取费用,费用根据咨询的范围而有所不同,目前费用为:51800欧元-103800欧元。
我们如何帮助您?
